Joint hypermobility syndrome, previously known as benign joint hypermobility syndrome, is a heritable disorder of connective tissue that presents with symptomatic hypermobility predisposing to arthralgia, soft tissue injury, and joint instability. It is indistinguishable from the hypermobility type of Ehlers-Danlos syndrome. Its complications can include autonomic dysfunction, chronic pain syndrome, proprioceptive impairment, premature osteoarthritis, dislocations, intestinal dysmotility, and laxity in other tissues causing hernias or uterine or rectal prolapse.
Joint hypermobility is very common, occurring in 10-20% of populations of Western countries, and higher in India, China, and Middle Eastern countries. It is important to distinguish between joint hypermobility and joint hypermobility syndrome. People who have hypermobile joints without symptoms are people with hypermobility. Those with symptoms attributable to their hypermobile joints may have joint hypermobility syndrome if they satisfy the Brighton criteria.


In a recent survey among members of the Hypermobility Syndrome Association, 52% of 251 patients waited over 10 years from the onset of symptoms to get a correct diagnosis.
If joint hypermobility syndrome is missed, the following problems may arise:
- Inappropriate and potentially harmful diagnosis or treatments such as rheumatoid arthritis, hypochondriasis, or somatisation.
- Excessive physical manipulation may cause avoidable damage, such as (a) precipitating subluxation or dislocation of intervertebral or peripheral joints, (b) rupture on ligaments, joint capsules, muscles, or tendons, or (c) precipitating pathological fractures in fragile bone. Exercise therapy may be either excessively forceful or ineffective.
Chronic pain may sometimes lead to immobility, deconditioning, dependency, and depression.
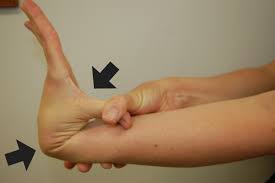
Diagnosis of hypermobility syndrome is clinical as currently no blood or imaging markers are available to confirm the diagnosis. The joint symptoms are secondary to a vulnerability to injury of fragile collagenous tissues (tendons, ligaments, muscles, bones, cartilages, and skin).
.
.
.
..
.
.
Management of the hypermobility syndrome:
Physician’s role in making a correct a diagnosis:
- To establish an accurate diagnosis. Marfan’s syndrome, or other forms of Ehlers-Danlos syndrome, such as vascular should be kept in mind. A positive family history of sudden early death from aortic aneurysmal dissection and/or rupture should suggest the possibility of Marfan’s syndrome, and a history of major spontaneous arterial rupture or uterine rupture in childbirth should raise suspicions of the vascular type of Ehlers-Danlos syndrome.
- Assessment of the musculoskeletal system, systemic involvement (dysautonomia, gastrointestinal dysmotility), declining mobility, and quality of life.
The role of physiotherapist is to help with:
- Core and joint stabilising and proprioception enhancing exercises.
- General fitness training.
- The use of techniques to restore mobility of joints or spine that was lost as a result of deconditioning and kinesiophobia (fear of movement).
- Beighton score of ≥4 (either currently or previously).
- Arthralgia for longer than three months in four or more joints.
Minor criteria
- Beighton score of 1, 2, or 3 (0, 1, 2, or 3 if aged > 50 years).
- Arthralgia in one to three joints or back pain or spondylosis, spondylolysis and/or spondylolisthesis.
- Dislocation in more than one joint or in one joint on more than one occasion.
- Three or more soft tissue lesions (eg, epicondylitis, tenosynovitis, bursitis).
- Marfanoid habitus (tall, slim, ratio of span to height greater than 1.03 and/or ratio of upper segment to lower segment less than 0.89, arachnodactyly).
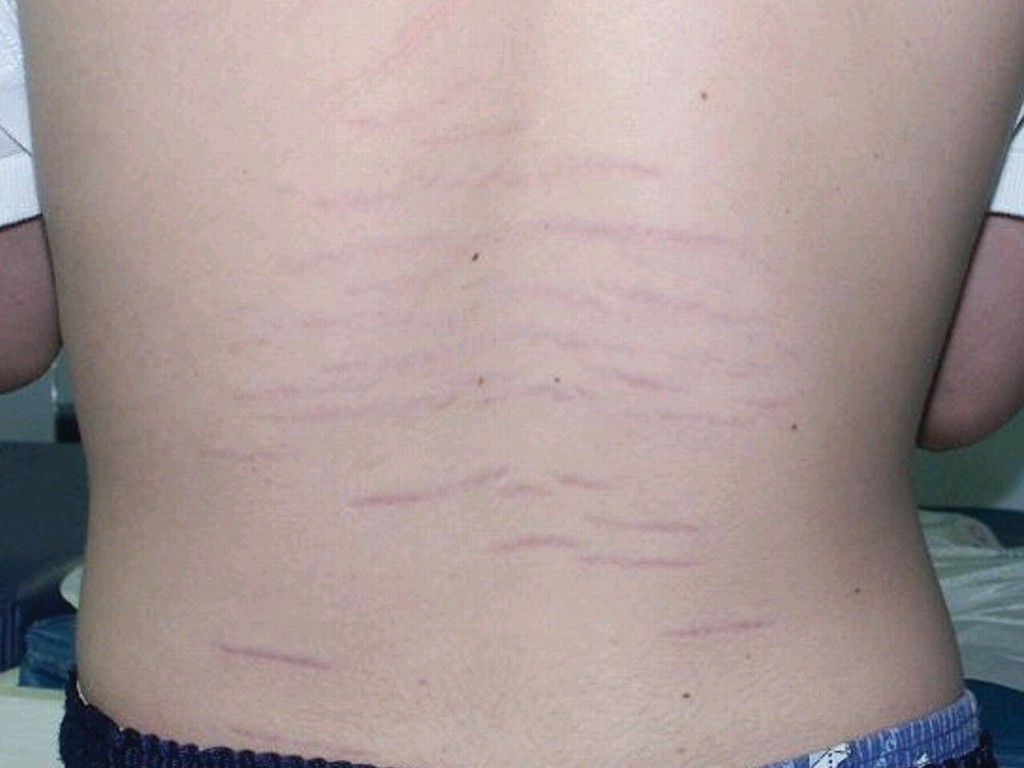
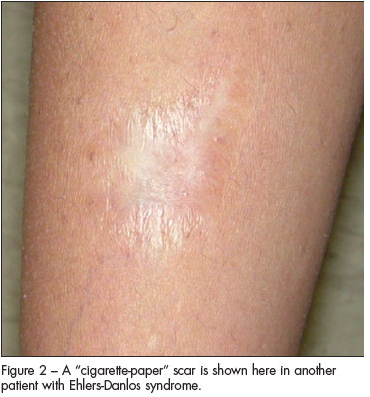
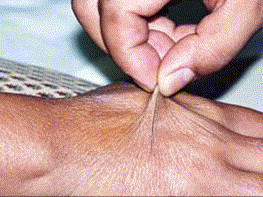
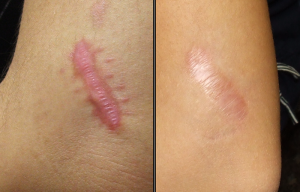
- Abnormal skin: striae, hyperextensibility, thin skin, papyraceous scarring.
- Eye signs: drooping eyelids, myopia, or antimongoloid slant.
- Varicose veins, hernia, or uterine or rectal prolapse.
.
This classification is old and was replaced recently by a new classification (not shown here).
The Beighton score measures a person’s degree of hypermobility. Score of 4/9 is considered positive. One point is assigned for the ability to accomplish each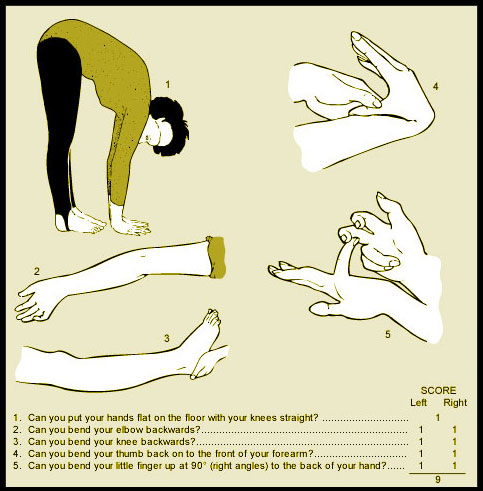
of the following movements:
- bending your small finger back further than 90 degrees (1 point each side)
- bending your thumb forward to touch your forearm (1 point each side)
- hyper-extending your elbows and knees, that is bending them beyond astraight line (1 point each joint, each side)
- putting your palms flat on the floor without bending your knees (1 point).
“A quide to living with hyper mobility syndrome” book by Isobel Knight
Singing dragon, ISBN: 9781848190689
http://www.bowen-technique.co.uk/pdfs/article032.pdf
.
.
“You know that you have hypermobility syndrome when…”, “You know that you have POTS when…” (books by Hannah Ensor)
www.stickmancommunications.co.uk
.
.
You may leave your email address to the “Your comments on the website content” and will be connected with other patients with hypermobility syndrome if more than one respond.
